


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
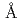 resolution
resolutionChatake, Toshiyuki*; Kurihara, Kazuo; Tanaka, Ichiro*; Tsyba, I.*; Bau, R.*; Jenney, F. E. Jr.*; Adams, M. W. W.*; Niimura, Nobuo
Acta Crystallographica Section D, 60(8), p.1364 - 1373, 2004/08
Times Cited Count:34 Percentile:88.87(Biochemical Research Methods)A neutron diffraction study has been carried out at 1.6 resolution on a mutant rubredoxin from 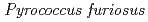 using the BIX-3 single-crystal diffractometer at the JRR-3 reactor of JAERI. In order to study the unusual thermostability of rubredoxin from
using the BIX-3 single-crystal diffractometer at the JRR-3 reactor of JAERI. In order to study the unusual thermostability of rubredoxin from 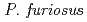 , the hydrogen-bonding patterns were compared between the native and a 'triple-mutant' variant where three residues were changed so that they are identical to those in a mesophilic rubredoxin. In the present study, some minor changes were found between the wild-type and mutant proteins in the hydrogen-bonding patterns of the Trp3/Tyr3 region. The H/D-exchange ratios in the protein were also studied. The results suggest that the backbone amide bonds near the four Cys residues of the FeS
, the hydrogen-bonding patterns were compared between the native and a 'triple-mutant' variant where three residues were changed so that they are identical to those in a mesophilic rubredoxin. In the present study, some minor changes were found between the wild-type and mutant proteins in the hydrogen-bonding patterns of the Trp3/Tyr3 region. The H/D-exchange ratios in the protein were also studied. The results suggest that the backbone amide bonds near the four Cys residues of the FeS redox center are most resistant to H/D exchange. In addition, the 1.6 resolution of the present neutron structure determination has revealed a more detailed picture than previously available of some portions of the water structure, including ordered and disordered O-D bonds.
redox center are most resistant to H/D exchange. In addition, the 1.6 resolution of the present neutron structure determination has revealed a more detailed picture than previously available of some portions of the water structure, including ordered and disordered O-D bonds.
by BIX-3, a single-crystal diffractometer for biomacromoleculesKurihara, Kazuo; Tanaka, Ichiro*; Chatake, Toshiyuki*; Adams, M. W. W.*; Jenney, F. E. Jr.*; Moiseeva, N.*; Bau, R.*; Niimura, Nobuo
Proceedings of the National Academy of Sciences of the United States of America, 101(31), p.11215 - 11220, 2004/08
Times Cited Count:48 Percentile:61.07(Multidisciplinary Sciences)The structure of a rubredoxin (Rd) from , an organism that grows optimally at 100  C, was determined using the neutron single-crystal diffractometer for biological macromolecules (BIX-3) at the JRR-3 reactor of JAERI. Data were collected at room temperature up to a resolution of 1.5 , and the completeness of the data set was 81.9 %. The model contains 306 H atoms and 50 D atoms. A total of 37 hydration water molecules were identified. The model has been refined to final agreement factors of
C, was determined using the neutron single-crystal diffractometer for biological macromolecules (BIX-3) at the JRR-3 reactor of JAERI. Data were collected at room temperature up to a resolution of 1.5 , and the completeness of the data set was 81.9 %. The model contains 306 H atoms and 50 D atoms. A total of 37 hydration water molecules were identified. The model has been refined to final agreement factors of 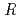 = 18.6 % and
= 18.6 % and  = 21.7 %. Several orientations of the O-D bonds of side chains, whose assignments from X-ray data were previously ambiguous, were clearly visible in the neutron structure. While most backbone N-H bonds had undergone some degree of H/D exchange throughout the molecule, five H atom positions still had distinctly negative (H) peaks. The neutron Fourier maps clearly showed the details of an extensive set of H bonds involving the ND
= 21.7 %. Several orientations of the O-D bonds of side chains, whose assignments from X-ray data were previously ambiguous, were clearly visible in the neutron structure. While most backbone N-H bonds had undergone some degree of H/D exchange throughout the molecule, five H atom positions still had distinctly negative (H) peaks. The neutron Fourier maps clearly showed the details of an extensive set of H bonds involving the ND
 terminus that may contribute to the unusual thermostability of this molecule.
terminus that may contribute to the unusual thermostability of this molecule.
Niimura, Nobuo; Kurihara, Kazuo; Tanaka, Ichiro
Kagaku, 59(2), p.46 - 47, 2004/02
no abstracts in English
